



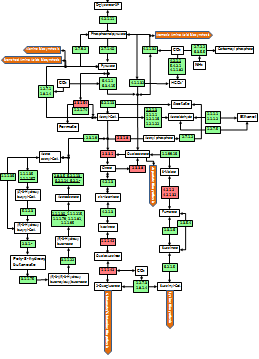
Metabolic and Functional Reconstruction
By comparing the names of a function listed in both databases, we can compile a subset of pathways that is the best fit for the current genome. This subset appears to be easy to compute automatically as an intersection of two sets of names: one being a list of all functions found in the current genome; the second - a list of all functions in our pathway database.
Depending on the type of pathway, the number of its reactions and catalytic and regulatory proteins involved, "the best fit" can be computed using different numeric criteria. The simplest is just the number of a particular pathway's proteins found in the genome as a percentage of the total number of proteins enlisted for the pathway. A more sophisticated way involves weighted sums of the found and enlisted proteins which would reflect relative contributions of particular proteins to the pathway functions. Signature proteins should have the highest weights, while unspecific enzymes (like unspecific enzymes attacking a chemical bond or an atom group) should be counted with a low weight because they can participate in a number of different pathways.
Besides sequence data, metabolic reconstructions can be devised from a transcriptome/ proteome or metabolome, alone or in any combination with each other.
Our sequence-based metabolic reconstruction technology predicts metabolic and functional potential of a cell corresponding to a fully expressed genome. Augmented with transcriptome, proteome and metabolome data, it allows for computing a much more specific or selective metabolic and functional model of cellular machinery.
The Process of Functional Reconstruction
The task of creating functional reconstructions from genome data can be broken into nine steps. Depending on the level of annotation of the input data, some steps may be unnecessary. However, re-running of certain procedures may enrich or invalidate some pre-existing data and thus may be valuable.
- Identification of genes
- Identification of operational subset of genes
- Functional assignment based on protein structure
- Initial assertion of pathways; identifying reconstruction outline
- Identification of missing genes
- Finding missing genes (filling functional vacancies)
- Finding missing functions (functions for vacant genes)
- Asserting additional pathways; repeating from step 5 if necessary
- Checking functional integrity, identifying possible phenotypes
Detailed information on each of these steps is available in a separate document upon request.
Email us for more information.