



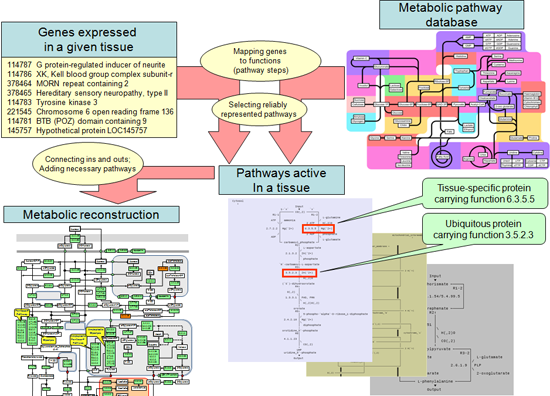
Reconstruction of Tissue-Specific Metabolism
In our tissue metabolism reconstruction process we are using technology originally developed for bacterial genome-based functional reconstruction, adjusted for transcriptomics data as the input. The process starts with automatic selection of pathways whose activity in the tissue is confirmed by the presence of corresponding transcripts.
We automatically categorize selected pathways by percentage of reactions for which the catalysts are evidently expressed. These categorized pathway sets constitute the raw reconstruction, while fully supported pathways comprise the skeleton of the reconstruction. Then the skeleton is updated by recruiting other pathways among partially supported by the expression data (or even completely unsupported). During this stage we use criteria based on the ontology of metabolic processes:
- Compositional constraint: the entire intermediary metabolism relevant to the tissue/compartment should be supported. This includes mandatory parts (like tissue-specific versions of the carbohydrate metabolism, oxidative phosphorylation or amino acid biosynthesis), semi-ubiquitous parts (like nucleotide biosynthesis in proliferating cells) and cell-type-specific parts (like bile acid biosynthesis in liver).
- Connectivity constraint: the resulting network should be fully connected, including the means for transport between cellular compartments and through external plasma membrane and means for the accumulation/retrieval of terminal compounds; and
- Parsimony principle: from all alternative sub-network sets, one containing the smallest number of unsupported reactions is used.
Simple following of these principles is not always possible, because often the choice should be made from equally unsupported sets of pathways. In such cases the choice is dictated by revisiting and careful analysis of the transcript sets, and sometimes even the genome. There are a number of reasons why the expression of a gene corresponding to an active function is not detected:
- the expression level could be too low, which often happens when the enzyme has favorable kinetics (low Km/high Vmax), so that even a very low concentration of the enzyme supports the necessary reaction rate; or when the corresponding protein is very stable;
- the corresponding transcript is improperly attributed to a gene (for instance, due to an unusual splice form);
- the gene is mis-annotated or not annotated;
- or the transcript is present, but the corresponding gene is unknown.
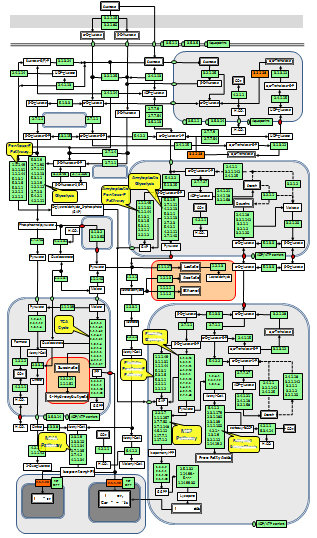
Considering all these cases and analyzing the evidences is a rigorous process requiring supervision by a highly trained expert. A diagram of the reconstruction process is presented below.
Please contact us for more information.